
Block copolymers are polymeric materials prepared by co-polymerizing different polymer monomers into chains, in which the different polymer monomers are arranged in a fixed order on a continuous chain [1,2]. Due to the special interactions between the segments, block copolymers are capable of self-assembling into nanoscale structures with specific morphologies, which have many applications in fields such as bioengineering and nanoscience. Typical examples of block copolymers include block olefin copolymers, block sulfoxide copolymers, and block polyester copolymers [3-6].
Thin-film materials are the precursor conditions for the development of microelectronics technology and the material basis for the manufacture of microelectronic devices. In the past half century, with the great progress of various film-forming methods, semiconductor thin-film materials have emerged from the body monocrystalline to amorphous, from amorphous to nano-phase, from narrow forbidden bands to wide bandgaps, and from conventional preparation to artificial design, a large number of new materials of high quality and important practical value [7-10]. At present, the study of semiconductor thin-film materials physics and technology has become an extremely active field in vacuum, microelectronics and materials science. The core of semiconductor thin-film materials research is the study of new materials and the improvement of the performance of traditional materials [11-14]. The former is the conception of new structural forms and the design of new chemical components according to the human will, and realized by modern ultra-thin layer epitaxy technology [15-17]. The latter is to change the microstructure of the material by using suitable process methods, so that it presents a new atomic configuration that conventional materials do not have [18,19].
Micro- and nanoscale honeycomb porous films have unique advantages, such as homogeneous pore size, high roughness, etc., and have a wide range of applications in the fields of separation membranes, optoelectronic devices, microreactors, template materials and superhydrophobic surfaces. In this paper, on the basis of combing the properties of block copolymers, semiconductor block copolymers were synthesized by introducing ATRP technology, and PEO-b-PMMA-b-PS honeycomb microporous membranes were prepared by combing the breath patterning method, and the morphological structure, thermal stability and optoelectronic properties of PEO-b-PMMA-b-PS honeycomb microporous membranes were analysed by scanning electron microscopy, heating and Hall tester. The PEO-b-PMMA-b-PS honeycomb microporous membrane was then used as a template, and the hollow open SnO\({}_{2}\) spherical particles were prepared by one-step blending method in combination with SnCl\({}_{4}\)·5H\({}_{2}\)O solution, and the corresponding SnO\({}_{2}\)-PS gas sensors were designed and experimentally analysed for their morphological structure and gas-sensitive properties.
Breath patterning is a method of constructing ordered honeycomb porous films using condensed and self-organised ordered water droplets as templates, which is convenient, fast, inexpensive, and the water droplets used as templates can be removed by natural evaporation. The pore size can be easily regulated by changing the relevant experimental parameters, and thus it has received a lot of attention in recent years. Honeycomb microporous films were prepared with the support of semiconductor block polymer materials in combination with the breath patterning method to provide a basis for analysing the application of honeycomb microporous films.
Block copolymers (BCPs) are a class of polymers in which polymers with different properties are linked by covalent bonds. Block copolymers can be classified into two-block copolymers, three-block copolymers, etc. based on the type of monomers constituting the block copolymers, and into linear block copolymers, dobby block copolymers, star block copolymers, etc. based on their topological structure. The structure of block copolymers affects their physical properties and phase separation morphology.
Usually, when two structurally different polymers are mixed, macroscopic phase separation occurs due to their thermodynamic incompatibility. Block copolymers can only undergo micro phase separation due to the covalent bonding between the different blocks. In the phase domain formed by microphase separation, the different blocks repel each other, and microphase separation structures with sizes ranging from 5 to 100 nm can be formed. The morphology and size of the microphase separation structures are related to the Flory Huggins interaction parameters, degree of polymerisation and volume fraction of the block copolymer.
At equilibrium, the microphase separation morphology of the bulk phase of the block copolymer is usually controlled by the primary structure of the block copolymer (e.g., chemical structure of the block, degree of polymerisation, volume fraction, etc.). In contrast, the microphase separation morphology of block copolymer films is also controlled by the surface free energy and interfacial free energy of the films.
The atom transfer radical polymerisation (ATRP) method is mainly based on simple organic halides as initiators and transition metal complexes as halogen atom carriers, and the control of the polymerisation reaction is achieved by establishing a dynamic equilibrium between active and dormant species through redox reactions . The specific mechanism is shown in Figure 1, where R-X are haloalkanes, \(M_{t}^{n} ,M_{t}^{n+1}\) is the reduced and oxidised transition metal, respectively, Ligand is the ligand, \(k_{a} ,k_{d}\) is the activation and deactivation reaction rate constants, respectively, \(k_{p}\) is the growth rate constant and \(k_{t}\) is the termination rate constant.

The following is an example of the ATRP reaction initiated by the RX/CuX/bpy system (where RX is a haloalkane and CuX is a cuprous halide) to further elaborate the basic principles of ATRP. The details are as follows:
Initiation stage \[\ R-X+CuX/bpy\to R\cdot +CuX2/bpy(X=C1Br)R\cdot +monomer\to P1\cdot . \tag{1}\]
Growth phase \[\ Pn-X+CuX/bpy\to Pn\cdot +CuX2/bpy(X=C1Br)Pn\cdot +monomer\to Pn+1\cdot . \tag{2}\]
Termination phase \[\ Pn\cdot +Pm\to Pn+mor\left(PnH+PmH\right) . \tag{3}\]
During the initiation phase, CuX and bpy complexes in the low oxidation state take halogen atoms from R·X to generate the primary radical R- and the dormant species of CuX2/bpy complexes in the high oxidation state. The primary radical then initiates the monomer to generate the monomer radical, the active species. The active species can either continue to initiate monomers for active polymerisation or take halogen atoms from the dormant species and become dormant itself. The preparation of structurally controllable polymers by polymerisation of “reactive” radicals requires a low steady-state concentration of chain-growing radicals, and the key lies in the establishment of a fast dynamic equilibrium between the dormant and reactive species (Pn-X+CuX/bpy and Pn·+CuX2/bpy).
This polymerisation reaction is called atom transfer polymerisation because the reversible transfer in this polymerisation reaction involves the atom transfer process of halogen atoms from halides to metal complexes and then from metal complexes to free radicals. At the same time, it is called atom transfer radical polymerisation (ATRP) because the reactive species are radicals.
Copolymer templating agents are mainly synthesised using anionic/cationic polymerisation techniques and reactive radical polymerisation (CRP) techniques, which in turn include atom transfer radical polymerisation (ATRP), reversible addition-fracture chain transfer polymerisation (RAFT) and initiating transfer terminators. The synthesis and application of block copolymer templating agents provide more templating agent options for the development of ordered porous materials.
ATRP synthesis technique is widely used to design and synthesise amphiphilic block copolymer templating agents with ultra-high molecular weights and high glass transition temperatures due to its ease of handling and high tolerance of impurities in the synthesis system. In this paper, taking PEO-b-PMMA-b-PS as an example, the polymerisation of methyl methacrylate monomer was initiated by using the macromolecular initiator PEO-Br to synthesize ultra-high molecular weight PEO-b-PMMA, based on which the more complex PEO-b-PMMA-b-PS semiconductor block copolymer templates were synthesized further. The specific formation process is shown in Figure 2.

The synthesis process of PEO-b-PMMA-b-PS is specified as follows:
By using the macromolecular initiator PEO-Br with different molecular weights (2000\(\mathrm{\sim}\)10,000 g/mol) and controlling the polymerisation time of methyl methacrylate (MMA), the block copolymer templates with different PEO chain lengths and different PMMA chain lengths (1\(\mathrm{\times}\)10\({}^{4}\)\(\mathrm{\sim}\)5\(\mathrm{\times}\)10\({}^{4}\) g/mol) were obtained. The polymerisation of styrene was further initiated with PEO-b-PMMA containing bromine at the end group to synthesise a more gradient hydrophilic and hydrophobic three-component triblock PEO-b-PMMA-b-PS semiconductor block copolymer template agent.
There are two general methods for preparing ordered porous films by breath patterning method, dynamic breath patterning method and static breath patterning method. Breath patterning method to prepare the texture is shown in Figure 3. Dynamic breath patterning is the process of preparing a porous film by allowing a continuous flow of humidity through the surface of the polymer solution, while static breath patterning is the process of forming a porous film by first creating an environment with a certain humidity atmosphere and then placing the polymer solution in this atmosphere.
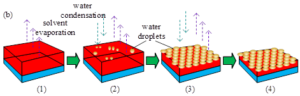
It is now generally accepted that honeycomb porous membranes are prepared as follows:
The polymer is dissolved in an organic solvent with a low boiling point and insoluble in water (commonly trichloromethane or carbon disulphide), and the solvent evaporates rapidly to produce a certain temperature gradient.
The surface temperature of the polymer solution decreases dramatically, leading to condensation of water vapour in a high humidity environment and the formation of small droplets of nanometre diameter on the surface of the solution, a process known as rapid nucleation of the droplet template. In this process, the growth of the diameter of the droplets with time conforms to equation \(D\approx t^{\alpha }\), where \(\alpha \approx 1/3\).
As the volatilisation-condensation process continues, the solution’s own temperature gradient induces Marangoni convection, the droplets grow and self-assemble into ordered arrays, and the droplet diameter grows with time in accordance with equation \(D\approx t^{\alpha }\), where \(\alpha \approx 1\). The droplets are covered by the polymer during this process, which avoids agglomeration of droplets with each other and ensures homogeneity of droplet size.
After complete volatilisation of solvent and water, honeycomb-like ordered array of pores is formed.
This time, the preparation of semiconductor block copolymer PEO-b-PMMA-b-PS honeycomb microporous film of the preparation of drugs is mainly as follows:
Semiconductor block copolymer PEO-b-PMMA-b-PS solution, previously homemade. Dichloromethane, trichloromethane, N methyl pyrrolidone, sodium chloride, potassium chloride, potassium nitrate, concentrated sulfuric acid, sodium hydroxide, of which concentrated sulfuric acid is 98% diluted to 10%, and the above pharmaceuticals were purchased from the National Pharmaceutical Group Corporation, and also homemade deionised water.
The main instruments used in this preparation of semiconductor block copolymer PEO-b-PMMA-b-PS honeycomb microporous membrane were as follows:
Electronic analytical balance (FAI205), purchased from SH Ke Tianmei Instruments, CNC ultrasonic cleaner (KQ-300), purchased from KS Ultrasonic Instrument Company, electric constant temperature blast drying oven (DHG-900), purchased from SH Yuezhong Instrument Company, magnetic heating stirrer (CJJ79), purchased from SH Meichun Instrument Factory, digital display constant temperature water bath (HH-2), purchased from JT City Chengdong Instrument Factory, low-temperature coolant circulation pump (DLSB), purchased from ZZ Great Wall Science and Industry Trade, optical microscope (BH12-UMA), purchased from O Company, SEM scanning electron microscope (Nova Nano), purchased from SH Yuzhong Industry.
A certain amount of PEO-b-PMMA-b-PS solution was dissolved in dichloromethane, trichloromethane, N methyl pyrrolidone solvents, respectively, and formulated into a concentration of chemically 0.6wt%\(\mathrm{\sim}\)2.2wt% of the casting solution, and defoamed using ultrasonic apparatus after solubilisation by range mixing, after a period of static time, and then left to stand for use after complete defoaming. The static method process is to prepare a saturated salt solution of certain humidity in a closed container, in which the saturated solutions of sodium chloride, KCL and potassium nitrate represent different humidity at different temperatures. The closed container is placed in a constant temperature water bath, and the prepared casting solution is dripped onto a slide, which is placed in the closed container and slowly formed into a film with the gradual evaporation of the solvent. Dynamic method is mainly in the adjusted humidity, the film casting liquid written casting on the slide, the adjusted smooth air flow into the surface of the film casting liquid, to wait for the solvent and water volatilisation is complete, that is, the honeycomb structure of the porous membrane.
Testing of the morphology of honeycomb microporous membranes
The surface morphology of PEO-b-PMMA-b-PS honeycomb microporous films prepared by the breathing pattern method was observed using a polarised light microscope and a tungsten filament scanning electron microscope. The glass sheet carrying the polymer honeycomb microporous film was cut and broken from the back side using a glass cutter, and the film was also broken at the corresponding position, and the cross-sectional morphology of the porous film was observed using a tungsten filament scanning electron microscope.
Characterisation of the thermal stability of honeycomb microporous films
The PEO-b-PMMA-b-PS honeycomb microporous membranes were placed on a hot table, which was raised to 140 \(\mathrm{{}^\circ}\)C at a rate of 20 \(\mathrm{{}^\circ}\)C/min and kept at 90 \(\mathrm{{}^\circ}\)C, 120 \(\mathrm{{}^\circ}\)C and 140 \(\mathrm{{}^\circ}\)C for 3 h. The changes of the honeycomb microporous membranes were observed during the heating period, and the PEO-b-PMMA-b-PS honeycomb microporous membranes heated to different temperatures were removed using a SEM. to observe the morphology of the porous membrane.
Characterisation of electrical properties of honeycomb microporous membranes
In this experiment, the electrical conductivity of the PEO-b-PMMA-b-PS honeycomb microporous film was tested by using the HMS-300 Hall tester manufactured by E. The test results of this test device contain parameters such as carrier concentration, carrier mobility, resistivity, square resistance, and Hall coefficient of the film. The test device makes use of the Hall properties of the material and measures each electrical parameter of the film under the action of an applied magnetic and electric field. The device is equipped with a magnet with a magnetic field strength of 0.55 T. The size of the sample is 8 mm \(\mathrm{\times}\) 8 mm. Before testing, indium metal electrodes need to be made at the corners of the sample to guarantee full contact between the film and the sample probe. Before starting the test, the thickness of the film and the current need to be set, and the appropriate current value is selected according to whether the I-V curve of the film is straight or not. After determining the electrical parameters of the PEO-b-PMMA-b-PS honeycomb microporous films, the electrical properties of the honeycomb microporous films under stress changes were probed using vertical and horizontal stretching.
PEO-b-PMMA-b-PS honeycomb microporous membranes were prepared using the breath patterning method based on semiconductor block copolymers. This chapter focuses on the quantitative data analysis of the physicochemical properties of this microporous membrane to provide data support for the realisation of PEO-b-PMMA-b-PS honeycomb microporous membranes.
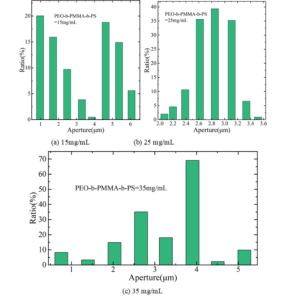
In this paper, honeycomb microporous membranes were prepared by mixing a mixture of NaCl, KCL, and KNO3 with semiconductor block copolymer PEO-b-PMMA-b-PS solution. In order to verify the effect of different concentrations of the semiconductor block copolymer solution on the structure of the honeycomb microporous membranes, the solution concentrations of 15 mg/mL, 25 mg/mL and 35 mg/mL were selected respectively, and the honeycomb microporous membranes obtained were observed by SEM and the corresponding pore size distributions were counted as shown in Figure 4. Among them, Figure 4(a)\(\mathrm{\sim}\)(c) shows the honeycomb pore size distribution at 15 mg/mL, 25 mg/mL, and 35 mg/mL concentrations, respectively.
As can be seen from the figure, when the semiconductor block copolymer PEO-b-PMMA-b-PS solution concentration is 15 mg/mL, the surface of the film shows a porous structure, and the pores are divided into larger pores and smaller pores, and most of the smaller pores are arranged at the bottom of the larger pores and on the connecting branches where the larger pores are connected to the larger pores. As a result, the pore diameter range is in two intervals, the average diameter of the larger holes is about 5.37 \(\mu\)m, and the average diameter of the smaller holes is about 1.69 \(\mu\)m.This is due to the fact that in the film-forming process, the semiconductor block copolymer will be deposited to the interface between the organic solvent and water to form a thin polymer film, and when the concentration of the semiconductor block copolymer solution is lower, the solution contains fewer semiconductor block copolymers, and the polymer film The surface strength is correspondingly weaker, leading to the formation of smaller pore structures at the bottom of the macropores and on the connecting branches as well. As the concentration of the semiconductor block copolymer solution was increased to 25 mg/mL, the polymer surface strength became stronger and the surface tension was subsequently reduced, effectively stabilising the water droplets. As a result, the formed film has a well ordered honeycomb structure with uniform and well aligned pore sizes, and the average diameter of the pores is about 2.85 \(\mu\)m (Figure 4(b)). When the concentration of the semiconductor block copolymer PEO-b-PMMA-b-PS solution continued to increase to 35 mg/mL, the volatilisation rate of the semiconductor block copolymer solution slowed down due to the large concentration of the solution. At the same time, the coalescence phenomenon occurred between some of the water droplets during film formation, and the porous film formed had a relatively large pore size and a wide range of distribution (Figure 4(c)). It can be seen that for the semiconductor block copolymer PEO-b-PMMA-b-PS solution system, the best pore structure regularity was obtained at a concentration of 25 mg/mL, and uneven or irregular pore structures were formed at too high or too low a concentration.
The scanning electron microscope described above shows the structural surface of the semiconductor block copolymer PEO-b-PMMA-b-PS honeycomb microporous membrane, and the structure of the prepared PEO-b-PMMA-b-PS honeycomb microporous membrane is characterised in this section by reflection infrared. As a reference, the untreated pristine semiconductor block copolymers were also tested for their IR properties. Figure 5 shows the IR characterisation results of the semiconductor block copolymer PEO-b-PMMA-b-PS honeycomb microporous membrane.
The IR profile of the pristine semiconductor block copolymer, where the absorption peaks between 3000 cm\({}^{-1}\)-3261 cm\({}^{-1}\) are attributed to the stretching vibrations of carbon-hydrogen bonds on the alkyl group and carbon-hydrogen bonds on the benzene ring, respectively. Whereas the absorption peak at 1678 cm\({}^{-1}\) is a characteristic absorption peak of the benzene ring skeleton, the absorption peaks at 1549 cm\({}^{-1-1}\)498 cm\({}^{-1}\) are the bending vibration absorption peaks of the C-H of the benzene ring. In addition, the strong absorption peaks between 750 cm\({}^{-1}\)-872 cm\({}^{-1}\) are the characteristic absorption peaks of the mono-substitution of the benzene ring. The infrared pattern of the honeycomb microporous membrane of the semiconductor block copolymer modified by the breathing pattern method adds several absorption peaks to the original pattern. The most obvious ones are the strong absorption peaks located between 1495 cm\({}^{-1}\)\(\mathrm{\sim}\)1253 cm\({}^{-1}\), which belong to the characteristic absorption peaks of P=N (1495 cm\({}^{-1-1}\)305 cm\({}^{-1}\)) and diphenyl sulfone (1279 cm\({}^{-1}\)), respectively. And the absorption peaks in the interval between 1318 cm\({}^{-1}\) and 1206 cm\({}^{-1}\) belonged to the vibrational absorption peaks of P-O-Ar formed by cyclotriphosphonitrile and BPS. From this, it is judged that we have successfully prepared the in situ polymerisation of PEO-b-PMMA-b-PS honeycomb microporous membrane surface based on semiconductor block copolymer and formed the corresponding nanostructures.

Based on the previous characterisation method of the thermal stability of PEO-b-PMMA-b-PS honeycomb microporous membranes, their thermal stability was analysed with respect to their thermal stability, with the expectation that their morphology can be kept undamaged during the application process and that their application range can be expanded. The PEO-b-PMMA-b-PS honeycomb microporous membranes were placed in a muffle furnace at three different temperatures, 90\(\mathtt{{}^\circ\!{C}}\), 120\(\mathtt{{}^\circ\!{C}}\) and 140\(\mathtt{{}^\circ\!{C}}\), and the PEO-b-PMMA-b-PS honeycomb microporous membranes were heated for 2h and then cooled down to room temperature and removed from the furnace, and then the morphology was observed by scanning electron microscopy, and the change of pore size of the PEO-b-PMMA-b-PS honeycomb microporous membranes was statistically determined at different temperatures. Figure 6 shows the pore size changes at different temperatures, where Figures. 6(a)\(\mathrm{\sim}\)(c) show the pore sizes at 90 \(\mathrm{{}^\circ}\)C, 120 \(\mathrm{{}^\circ}\)C and 140 \(\mathrm{{}^\circ}\)C, respectively.
As can be seen from the figure, when the temperature was heated to 90 \(\mathrm{{}^\circ}\)C, the porous structure of the PEO-b-PMMA-b-PS honeycomb microporous membrane still existed, and the average diameter of its micropores as a whole was in the range of 3.51 \(\mu\)m to 4.71 \(\mu\)m. The average diameter of the micropores further expands to 5.25 \(\mu\)m after the temperature is continuously increased to 120 \(\mathrm{{}^\circ}\)C. When the heating temperature is increased to 140 \(\mathrm{{}^\circ}\)C, the pore diameter of the PEO-b-PMMA-b-PS honeycomb microporous membrane has expanded to 12.42 \(\mu\)m\(\mathrm{\sim}\)14.13 \(\mu\)m, and at this time, the PEO-b-PMMA-b-PS honeycomb microporous membrane has changed from the original milky white to transparent. This indicates that the structure of the semiconductor block copolymer changes from rigid to viscous fluid as the temperature increases, and the overall structure of the PEO-b-PMMA-b-PS honeycomb microporous membrane tends to be in the stage of destruction. Therefore, when using PEO-b-PMMA-b-PS honeycomb microporous membranes with semiconductor block copolymers, it is necessary to pay attention to the control of the temperature of the experimental environment in order to obtain more usable honeycomb microporous membranes.

For the optical properties of PEO-b-PMMA-b-PS honeycomb microporous films, SEM scanning electron microscopy was mainly used to analyse the optical properties, in addition, the chloroform solution of MEH-PPV as well as the dense films prepared by the drop-coating method were chosen as comparisons to analyse the UV-visible absorption spectra of different types of honeycomb microporous films. Figure 7 shows the UV-visible absorption spectra. The X1 curve represents the chloroform solution of MEH-PPV, with the maximum absorption peak at 4850 cm\({}^{-1}\), generated by \(\Pi\)–\(\Pi\)* jump absorption.The X2 curve represents the dense thin film prepared by drop-coating method, with the maximum absorption peak at 4927 cm\({}^{-1}\) .The X3 curve represents the honeycomb porous structured thin film of PEO-b-PMMA-b-PS prepared by breath patterning method, with the maximum absorption peak at 5108 cm\({}^{-1}\). It can be seen that the absorption peaks of the polymer dense films (X2\(\mathrm{\sim}\)X3) are obviously broadened and slightly red-shifted, and the PEO-b-PMMA-b-PS honeycomb porous structure film has the widest spectral band, which is due to the change of the aggregation state.

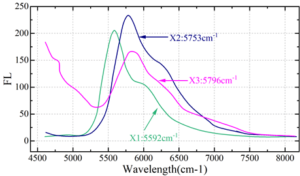
Figure 8 shows the fluorescence spectra at excitation wavelengths of 4500 cm\({}^{-1}\), where X1\(\mathrm{\sim}\)X3 are the same as above. The MEH-PPV chloroform solution shows a main peak at 5592 cm\({}^{-1}\) and a shoulder peak at 5996 cm\({}^{-1}\), and the dense film prepared by the drop-coating method shows a main peak at 5753 cm\({}^{-1}\) and a shoulder peak at 6217 cm\({}^{-1}\). In contrast, the semiconductor block copolymer PEO-b-PMMA-b-PS honeycomb microporous film prepared by the breath patterning method has a similar peak shape to that of X2, and the wavelength position of the maximum peak is not significantly shifted. That is, the peak positions of the fluorescence peaks of the solid films did not change significantly, whereas the peak positions of the fluorescence peaks of the dense and porous films had a redshift of about 161 cm\({}^{-1}\) compared with the MEH-PPV chloroform solution. This indicates that the luminescent properties of the porous films formed under high humidity environment are unchanged compared to the dense films, while the displacement of the fluorescence peaks is caused by the difference in the aggregation state of the polymer materials.
The results of characterisation by fluorescence spectroscopy and confocal fluorescence microscopy show that the semiconductor block copolymer PEO-b-PMMA-b-PS honeycomb microporous thin films prepared by the breath patterning method have good optical properties.
Better stretchability is usually obtained by combining organic semiconductors with elastomers. Pre-polymers of semiconductor block copolymers and cross-linkers were first spin-coated onto PEO-b-PMMA-b-PS films prepared using the breath patterning method, and then heated and cured in order to form the PEO-b-PMMA-b-PS bilayer structure. Subsequently, experiments were conducted using a small amount of hydrofluoric acid to etch the glass to peel the PEO-b-PMMA-b-PS bilayer from the glass substrate without causing damage to the film.
In order to investigate the change in electrical conductivity of oriented PEO-b-PMMA-b-PS films under strain stretching conditions, we tested the current-voltage curves of the films under different strains using a homemade microtensile machine in situ after doping the PEO-b-PMMA-b-PS films obtained using the above method, so as to further characterise the change in electrical conductivity. In this experiment, the PEO-b-PMMA-b-PS films were tested using the two-probe method, in which two probes were placed on two electrodes along the direction of the long axis of the PEO-b-PMMA-b-PS crystal fibre bundles to test the changes in the conductivity of the films. At the same time, the samples were stretched, and one strain value was fixed at a time for testing, and there were two kinds of stretching directions, one was perpendicular to the direction of the fibre bundles of the PEO-b-PMMA-b-PS films, and the other was along the direction of the long axis of the fibre bundles. Figure 9 shows the conductivity test results of the PEO-b-PMMA-b-PS films, where Figures 9(a)\(\mathrm{\sim}\)(b) show the test results of the perpendicular and parallel directions, respectively.
By stretching the PEO-b-PMMA-b-PS films in the vertical direction and retrograde, initially, the current does not change much with the increase of strain at smaller tensile stresses, which shows that the PEO-b-PMMA-b-PS films are able to maintain their relative integrity and stability under smaller stretching effects. Subsequently, when the strain is increased to 60%, the current density gradually decreases with increasing strain, at which point the films still have some conductive stability. The current density does not decrease significantly until the strain reaches over 100%. When the PEO-b-PMMA-b-PS films were stretched parallel to the direction of the fibre bundle, the initial current was relatively stable at less than 20% strain during the pre-stretching process. Subsequently, the current density decreased significantly at strains above 40%. After the strain exceeds 60%, the current density decreases significantly, which indicates that the PEO-b-PMMA-b-PS film has caused serious damage.

In order to quantitatively describe the trend of the electrical conductivity of the PEO-b-PMMA-b-PS films, the magnitude of the corresponding conductivity at each tensile strain was further calculated by a Hall tester. Figure 10 shows the relationship between the conductivity of PEO-b-PMMA-b-PS films with strain intensity under two directions of tensile conditions.
The initial conductivity value of the semiconductor block copolymer PEO-b-PMMA-b-PS films prepared by the breath-patterned method after doping reaches more than 3.5 S/cm, which is significantly higher than that of the PEO-b-PMMA-b-PS films prepared directly by drop-coating (around 0.94 S/cm). The above indicates that the orientation arrangement of the molecular chains is beneficial to improve the electrical conductivity of the conjugated polymer films. For the PEO-b-PMMA-b-PS films, the change in conductivity clearly depends on the direction of the force applied to the films. When the films were stretched along the parallel direction of the PEO-b-PMMA-b-PS molecular chains, a sharp decrease in conductivity occurred at strain intensities above 40%. In contrast, when the films were stretched perpendicular to the direction of the PEO-b-PMMA-b-PS molecular chains, the decrease in conductivity was relatively small. When the strain is within 90%, the conductivity can still maintain a relatively high value (greater than 1 S/cm). When the film is stretched over 120% or more, the conductivity starts to decrease to 0.14 S/cm.The above results confirm that the semiconductor block copolymer PEO-b-PMMA-b-PS films prepared by the breathing pattern method exhibit a significant anisotropy of electrical conductivity under the tensile strain condition, and they also possess a relatively excellent electrical conductivity.

Currently, the research on semiconductor block copolymers is getting deeper and deeper, and functional micro- and nanomaterials of various sizes and morphologies have received extensive attention. The honeycomb microporous membrane of semiconductor block copolymers has a wide range of applications, and this chapter is mainly based on the PEO-b-PMMA-b-PS honeycomb microporous membrane prepared in the previous chapter, and the SnO\({}_{2}\) particles with hollow openings are prepared. The gas-sensitive properties are also analysed to provide a new research basis for the application of semiconductor block copolymer honeycomb microporous membranes.
On the basis of the experimental raw materials in the previous section, monodisperse SnO\({}_{2}\) nanoparticles were selected in this section, which were purchased from NJ East Inspection Biotechnology Company, ethanol was purchased from Sinopharm, and fibre-optic spectrometer (QE6500) was purchased from American O. NaHCO\({}_{3}\), SnCl\({}_{4}\)·5H\({}_{2}\)O, NH\({}_{3}\)·H\({}_{2}\)O, and HCl, the aforementioned reagents were purchased from Sinopharm Chemical Reagent Company. In addition, the semiconductor block copolymer PEO-b-PMMA-b-PS prepared as previously described was used as a template.
In this experiment, hollow SnO\({}_{2}\) spheres were prepared by a one-step blending method and coated onto 5 \(\mathrm{\times}\) 5 mm flat electrodes by combining PEO-b-PMMA-b-PS spheres and spin-coating to construct a porous thin-film-type NO\({}_{2}\) sensor (SnO\({}_{2}\)-PS), and the specific steps for the preparation of the sensitive materials and devices were as follows:
Preparation of SnO\({}_{2}\) spheres. PEO-b-PMMA-b-PS with a molecular weight of 1.5*10\({}^{5}\) was dissolved in CHCl3 and configured to a concentration of 15 mg/mL, and the solution SnCl\({}_{4}\)·5H\({}_{2}\)O was added to the solution in PEO-b-PMMA-b-PS/CHCl\({}_{3}\) in different proportions and then ultrasonically mixed, and formulated into a film-making solution for use. The SnCl\({}_{4}\)·5H\({}_{2}\)O/PEO-b-PMMA-b-PS/CHCl\({}_{3}\) solution was poured on the glass substrate under the condition of RH 30%\(\mathrm{\sim}\)80%, and the hollow SnO\({}_{2}\) balls were obtained after the solvent evaporated, and then used after vacuum drying at room temperature.
Fabrication of gas-sensitive sensor. Take equal volume of PEO-b-PMMA-b-PS ball solution, deionised water and anhydrous ethanol mixed according to the ratio of 1:1:1, ultrasonic dispersion uniform. Then the flat electrode was cleaned, firstly by ultrasonication in ethanol to remove surface impurities, after drying, the electrode was put into a vacuum plasma processor, and its surface was cleaned and hydrophilic treated with O\({}_{2}\) plasma. After the treatment of the electrode, 10 \(\mu\)L of the PEO-b-PMMA-b-PS ball mixture solution obtained earlier was taken and dripped onto the substrate, and then spun-coated with a homogeniser for 20 s at a speed of 2500 rpm, and then dried in an oven for 15 min at 100\(\mathrm{{}^\circ}\)C. After that, the SnO\({}_{2}\) nanosol was dripped-coated onto the substrate containing the PEO-b-PMMA-b-PS balls, and 4 \(\mu\)l was added to the substrate each time, and 3500 rpm was used for spin-coating. l, spin-coated at 3500 rpm for 20 s, and repeated 10 times, and after each spin-coating the substrate was placed on a heating table at 90\(\mathrm{{}^\circ}\)C for 12 min to cure the film layer. Finally, the PEO-b-PMMA-b-PS spheres capped by SnO\({}_{2}\) nanocrystals were removed by heating in a muffle furnace in two stages, the first stage with a heating rate of 3\(\mathrm{{}^\circ}\)C/min to 280\(\mathrm{{}^\circ}\)C for 2 h, and the second stage with a heating rate of 1.5\(\mathrm{{}^\circ}\)C/min to 450\(\mathrm{{}^\circ}\)C for 4 h. The substrates were also coated with PEO-b-PMMA-b-PS spheres in the same way (but without PEO-b-PMMA-b-PS). PMMA-b-PS spheres for making holes), the SnO\({}_{2}\) nanosol was directly spin-coated as a reference group to verify whether the use of PEO-b-PMMA-b-PS spheres for making holes enhanced the gas-sensitive characteristics of the sensor. The SnO\({}_{2}\)-PS gas sensor was obtained by soldering the fabricated flat electrodes to the tube holder for testing.
Structural characterisation of hollow SnO\({}_{2}\) spheres
XRD is an important analytical method to analyse the crystal structure and physical phase of materials. In this paper, the XRD analysis was carried out using a rotary-target X-ray diffractometer, model Rigaku D/max, under the conditions of Cu-target K\(\alpha\) line, voltage 50 kV, current 50 mA, continuous scanning, and scanning speed of 4 \(\mathrm{{}^\circ}\)/min. The XRD diffraction data were analysed by Jade software, and the SEM was mainly used to observe the surface and cross-section morphology of the material. In this paper, the morphology of the samples was observed with a JSM-6460 SEM to obtain the microstructure of the samples.The JSM-6460 SEM comes with an EDX spectrometer, which can be used to determine the elemental composition and content of the samples.
Gas-sensitive performance test
The gas-sensitive test instrument used in this paper is a homemade device. The model WS-30A gas-sensitive tester was utilised, which can provide heating voltage, fan and sealed container. The sample of SnO\({}_{2}\)-PS sensor with electrodes made was placed on the heated ceramic sheet and put into the sealed container. The sample is connected to the electrochemical workstation through a wire, and air is used as the equilibrium gas, and a certain concentration of the gas to be tested is injected into the sealed container with a syringe in accordance with the ratio, and the recording of the gas-sensitive signal is completed through the electrochemical workstation.
The gas-sensitive performance test parameters of the gas-sensitive sensor mainly include response value, response time and recovery time. Response value is an important indicator of the sensor’s standby characteristics, which is defined as the ratio of the output increment to the corresponding input increment. For gas-sensitive sensors, because its essence is the change of resistance, so, generally use the gas-sensitive response value to describe its response value, in this paper, the gas-sensitive response value is defined as the ratio of the resistance value when the sensor is in air and in a specific concentration of the atmosphere to be measured when it is stable. The time from the beginning of the passage of the gas to be measured until the signal output value of the gas-sensitive device changes to 80% of the maximum value is defined as the response time, and the time from the beginning of the passage of the air until the output signal is restored to 80% of the stable value in the original air atmosphere is defined as the recovery time.
Energy spectra of hollow SnO\({}_{2}\) spheres
The hollow SnO\({}_{2}\) spheres obtained by one-step blending method were prepared using the semiconductor block copolymer PEO-b-PMMA-b-PS honeycomb microporous membrane as a template, and their morphological features were observed by SEM. And the hollow SnO\({}_{2}\) spheres were sintered in a muffle furnace, heated up to 400 \(\mathrm{{}^\circ}\)C at 2 \(\mathrm{{}^\circ}\)C/min and kept at a constant temperature for 3 h, and then cooled naturally. After that, the heat treatment was continued for 3 h. After that, they were taken out to observe their morphology, and the energy spectra were compared and analysed before calcination, 3 h and 6 h later. Figure 11 shows the EDS diagrams of hollow SnO\({}_{2}\) spheres, where Figures 11(a)\(\mathrm{\sim}\)(c) show the results before calcination, after 3 h and after 6 h of calcination, respectively.
Before starting the calcination, it can be observed that the hollow SnO\({}_{2}\) spheres still maintain a better honeycomb porous structure, and the energy spectrum shows that Sn and Cl are the main elements of the porous film in addition to the C elements in the organic matter. After calcination for 3h, the porous membrane matrix was found to be partially destroyed, but it was observed that the hydrolysis products grew along the pore wall of the porous membrane under the structural guidance of the porous membrane, which still maintained relative order. The corresponding spectral analysis shows that the hollow SnO\({}_{2}\) spheres are mainly composed of Sn and O. This means that SnCl\({}_{4}\) has been completely hydrolysed and converted into SnO\({}_{2}\) during the heat treatment, and the matrix of the film has been completely destroyed and the hollow open-ended structural particles are fully unfolded after calcinations for 6 h. The porous film matrix has been partially destroyed, but it can be seen that the hydrolysis products grow along the porous film walls under the guidance of the porous film structure, which still maintains relative order.

XRD pattern of hollow SnO\({}_{2}\) spheres
Figure 12 shows the XRD spectrum of the product after calcination of hollow SnO\({}_{2}\) spheres. All the observed diffraction peaks are consistent with the standard spectrum of SnO\({}_{2}\) with tetragonal rutile structure, corresponding to the standard JCPDS card number NO40\(\mathrm{\sim}\)1459. The positions of the peaks are at 34.1\(\mathrm{{}^\circ}\), 41.8\(\mathrm{{}^\circ}\), and 62.3\(\mathrm{{}^\circ}\), which correspond to the (112), (113), and (209) crystal planes of tetragonal SnO\({}_{2}\), respectively. No other crystalline crystal phases were found. It indicates that the purity of the final product is very high, which further confirms that the hollow SnO\({}_{2}\) spheres satin-fired yielded tetragonal-phase rutile-structured inorganic oxides of SnO\({}_{2}\) with high purity.

In order to verify the gas sensitivity of the hollow SnO\({}_{2}\) spheres prepared in this paper using the semiconductor block copolymer PEO-b-PMMA-b-PS honeycomb microporous film as a template, a SnO\({}_{2}\)-PS gas sensor was designed based on the hollow SnO\({}_{2}\) spheres in this paper. For the gas sensitivity of this gas sensor, another pure SnO\({}_{2}\) film was prepared by hydrothermal method and a SnO\({}_{2}\) gas sensor was designed based on this film as a comparison. Figure 13 shows the gas-sensitive response test results, where Figures 13(a)\(\mathrm{\sim}\)(b) show the comparison results of the two gas sensors as well as the performance comparison of the gas sensors after drop-coating with different concentrations of PEO-b-PMMA-b-PS solutions, respectively.
The response of the simple SnO\({}_{2}\) gas sensor was compared and analysed with that of the SnO\({}_{2}\)-PS gas sensor in 2 ppm NO\({}_{2}\) gas at the same response/recovery time. The results showed that the response of SnO\({}_{2}\)-PS gas sensor was significantly improved, and the response reached -9.42% in 2 ppm NO\({}_{2}\) gas environment, which was 2.36 times higher than that of SnO\({}_{2}\) gas sensor.The response/recovery times of SnO\({}_{2}\) gas sensor and SnO\({}_{2}\)-PS gas sensor were 54/453s and 51/317s, respectively, and the response and recovery times were also significantly enhancement. The results of the SnO\({}_{2}\)-PS gas sensor response for drop-coated SnO\({}_{2}\)-PS gas sensors with different concentrations of PEO-b-PMMA-b-PS solution showed that the sensor response, although increased with the concentration of PEO-b-PMMA-b-PS, was not greatly enhanced.
PEO-b-PMMA-b-PS modification of SnO\({}_{2}\) to improve the gas response as well as the response/recovery time can be explained in the following two ways:
Chemical sensitisation effect. On the one hand, PEO-b-PMMA-b-PS modification increases the specific surface area and the number of adsorbed active sites of the SnO\({}_{2}\) material, which enables the transfer of NO\({}_{2}\) gas molecules to the surface of the SnO\({}_{2}\) material through the spillover effect. On the other hand, PEO-b-PMMA-b-PS acts as a catalyst material, and the modified SnO\({}_{2}\) can reduce the activation energy of the gas reaction at room temperature, which makes the gas molecules cleaved, and thus improves the reaction rate of the sensor.
Electron sensitisation: PEO-b-PMMA-b-PS alters the electronic structure or carrier concentration of SnO\({}_{2}\) through electron transfer with SnO\({}_{2}\), thus changing the gas-sensitive response performance of SnO\({}_{2}\).

In order to further analyse the gas-sensitive performance of the hollow-opening SnO\({}_{2}\)-PS gas sensor, continuity and repeatability tests were performed on the SnO\({}_{2}\)-PS gas sensor, and the gas-sensitive response test results of the SnO\({}_{2}\)-PS sensor are shown in Figure 14, where Figures 14(a)\(\mathrm{\sim}\)(b) show the continuity and repeatability test results, respectively.
The hollow-opening SnO\({}_{2}\)-PS gas sensors were tested for NO\({}_{2}\) gas with concentrations of 0.8 ppm, 1.2 ppm, 1.6 ppm, 2.0 ppm, and 2.4 ppm, and the responses were -6.53%, -11.34%, -17.26%, -21.55%, and -24.52%, which proved their good dynamic characteristics. Six consecutive cyclic tests were performed in 2 ppm NO\({}_{2}\) gas, and the amount of gas response was almost the same each time. During the recovery of the sensor, the resistance value is basically the same as the initial value, which proves that the hollow-open SnO\({}_{2}\)-PS sensor has good repeatability and reversibility.

In this article, semiconductor block copolymers were synthesised using ATRP, PEO-b-PMMA-b-PS honeycomb microporous membranes were prepared using the breath patterning method, and hollow-open SnO\({}_{2}\) spheres as well as SnO\({}_{2}\)-PS gas sensors were prepared in combination with a one-step blending method. For the PEO-b-PMMA-b-PS honeycomb microporous membrane, the best pore structure regularity was obtained when the concentration of PEO-b-PMMA-b-PS was 25 mg/mL, and its stability gradually decreased with the increase of temperature, and the stability was better when the temperature was lower than 140 \(\mathtt{{}^\circ\!{C}}\). For the hollow open-ended SnO\({}_{2}\) spheres, the diffraction peaks are located at 34.1\(\mathrm{{}^\circ}\), 41.8\(\mathrm{{}^\circ}\), and 62.3\(\mathrm{{}^\circ}\), which correspond to the (112), (113), and (209) crystal planes of tetragonal-phase SnO\({}_{2}\), respectively.The response of the SnO\({}_{2}\)-PS gas sensor has been significantly improved to -9.42% in a 2 ppm NO\({}_{2}\) gas environment, which is 2.36 times higher than that of the SnO\({}_{2}\) gas sensor by a factor of 2.36. The application of microporous honeycomb films prepared from semiconductor block copolymers to nanohollow SnO\({}_{2}\) spheres can effectively enhance their gas-sensitive properties and expand the application fields of microporous honeycomb films of semiconductor block copolymers.
Although the stability and reproducibility of the multi-cycle response of hollow-open SnO\({}_{2}\) gas sensors prepared by honeycomb microporous membranes of semiconductor block copolymers have been preliminarily investigated, their long-term stability in real environments needs to be further evaluated. In addition, the experimental results show that the honeycomb microporous membranes of semiconductor block copolymers have good electrical properties, and the conductive gas-sensitive response can be combined with Raman signals for more in-depth application studies in subsequent studies to further enhance the application of semiconductor block copolymers.